I included tutorials about access to ENCODE data, an Intro to Linux for complete computing novices (quite a few of our students), Genomic Intervals in the UCSC Genome Browser, use of BEDTools to compare genomic intervals for various factors, and an a tutorial in R for data display. Later in the week we looked at gene expression with RNA-seq using TopHat and Cufflinks. The general plan for the 5-day workshop (for 6 students) was as follows:
Monday
9-11:00 am Lecture (2 hr): Introduction to Gene
Regulation and Epigenetics
11-12:00 am Lecture (1 hr): Use of the HighPerformance Computing Cluster
12:00-2 pm Working Lunch with HPC System Manager
(2 hr): Set up HPC account for each student, practice Linux commands, move
files from laptop to HPC account
2-4 pm Exercise 1: Tutorials for Accessing ENCODE data through the ENCODEPortal, UCSC Genome Browser and ENSEMBL Browser
Tuesday
9-11:00 am Lecture & Demo: (2 hr): The UCSC
Genome Browser, BED file format, and BEDTools software
11-12:00 am Exercise
2: BEDTools Tutorial
12-1:00 pm Lunch
1-3:00 pm Exercise
3: Use of ENCODE Data and BEDTools to compute the Intersection of DNAse
hypersensitive sites with promoters of all RefSeq genes
Wednesday
9-10:30 am Lecture: Computing Gene Expression
with RNA-Seq (1.5 hr)
10:30-12 am
Exercise 4: Align ENCODE RNA-seq data to hg19 reference genome with TopHat
12-1:00 pm Lunch
1-4 pm Continue work on Exercise 4
Thursday
9-10:00 am Lecture (1 hr): Intro to data
visualization with R
10-12:00 am Exercise 5: TryRCodeschool tutorial.
12-1:00 pm Lunch
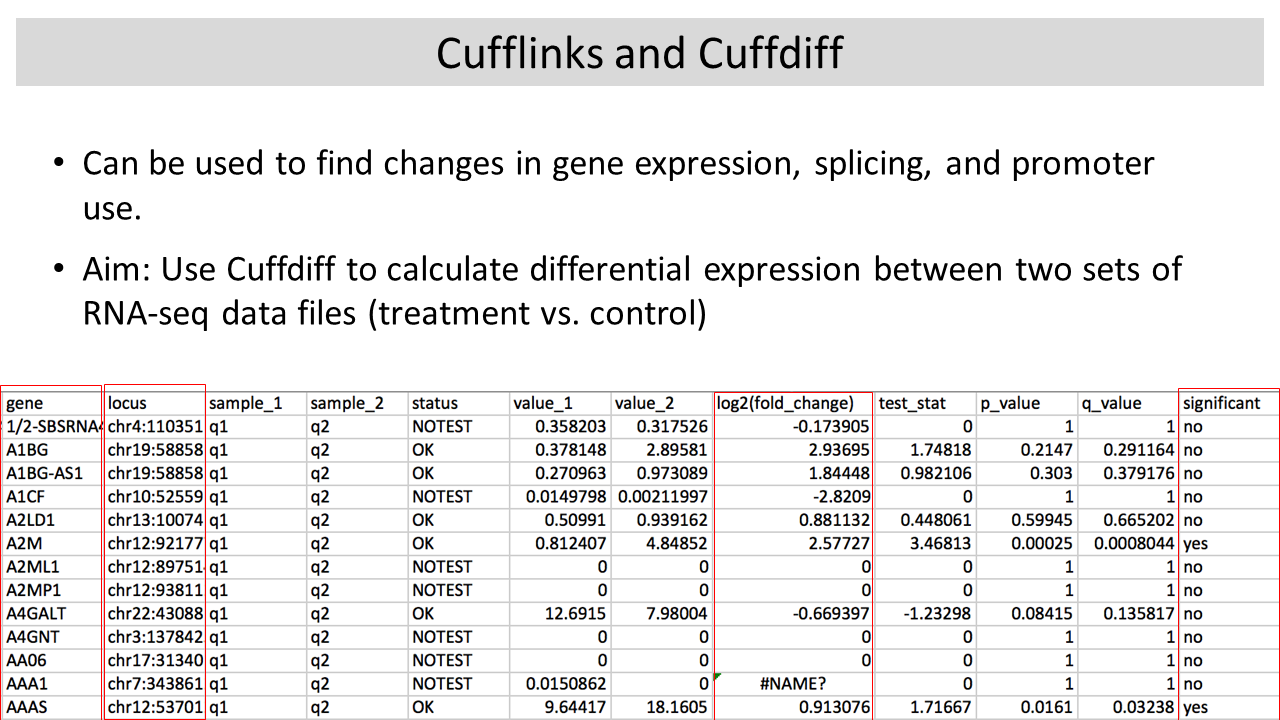
1-2:00 pm Lecture (1 hr): Differential Gene
Expression with Cufflinks
2-4:00 pm Planning
for Research Project – choose ENCODE data for transcription factors, gene
expression, and epigenetic markers. Literature search.
Friday
9-12:00 am Work
on Research Project
12-1:00 pm Lunch
1-4:00 pm Work
on Data analysis and prepare presentation
I had six students in our Research team: Elaine Fisher, Reuben Moncada, Shushan Sargsian, Beny Shapiro, Jong Shin, and Bo Xia, I have pasted images from their final presentation below (can't upload PowerPoint or PDF in this Blogger).
My overall impression of the week was that the students learned a huge amount of computing skills, but it was a bit bumpy when we got to the RNA-seq methods. They had really good success comparing various Transcription Factor binding sites to known genes (promoter region, TSS, 3'UTR, exons, introns, 5'UTR), finding interactions between TF's by finding overlapping or nearby binding sites, We also found nice overlaps between ChIP-seq TF binding sites and DNAse sensitive sites, histone modification sites, and computationally predicted TF binding sites. Also, the students did a nice job of measuring overlapping vs. nearby binding sites (bedtools slop), and measuring the significance of intersections using bedtools shuffle to create a statistical model of random intersections as a control.
FASTQ data download and alignment is slow and error prone (we had a lot of trouble making SGE scripts that would run correctly on our compute cluster). I should have shown TopHat just as a demo and used a small local FASTQ data file as an example rather than download and re-align ENCODE data. Using Cufflinks/Cuffdiff to compare gene expression from different cell lines was feasible with real ENCODE BAM files, but we had to learn this earlier in the week and spend more time to create SGE scripts that would run nicely with multithreading (to complete in a reasonable amount of time).
If I did this sort of tutorial again, I would figure out a way for the students to measure differential gene expression between cell lines from pre-computed ENCODE RNA-seq quantified data (wig files).

















{kind=link}